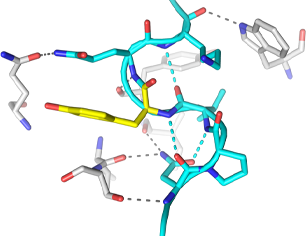

Protein X-ray Crystallography
Protein X-ray crystallography is a powerful technique used to determine the three-dimensional structure of proteins at atomic resolution. This technique allows researchers to map the arrangement of atoms within the protein, and reveal crucial information and insights into its structure, function, interactions, and potential as a target for drug development.
It involves crystallizing a purified protein, exposing the crystal to X-rays, and analyzing the diffraction patterns produced.
Protein Crystallization

Protein crystallization is the crucial first step in X-ray crystallography, where a purified protein sample is carefully combined with crystallization solutions to form crystals.
These crystals are essential because they create a repeating, lattice-like structure of protein molecules that are needed to generate diffraction patterns when exposed to X-rays. Achieving high-quality crystals is fundamental, as the quality of the resulting X-ray data directly depends on the precision and regularity of the crystals formed in this initial step.
The TCSB is equipped with cutting-edge robotics for protein crystallization, offering researchers access to advanced automated systems. These state-of-the-art tools enhance the efficiency and precision of crystallization experiments.
The facility's robotic platforms include:

Mosquito HTS, SPT Labtech
The Mosquito HTS is a highly advanced robot designed specifically for protein crystallization experiments. It automates the process of combining protein samples with various crystallization solutions, significantly improving the efficiency of crystallization trials.
One of its key advantages is its ability to dispense extremely small volumes, in the nanoliter range, which drastically reduces the amount of protein required for screenings. This is particularly valuable given the challenges of obtaining large quantities of pure protein. Additionally, the Mosquito HTS is highly versatile, supporting both hanging-drop and sitting-drop vapor diffusion methods, ensuring consistent and precise results across experiments.

Rock Imager, Formulatrix
The ROCK IMAGER 1000 is an automated system designed for high-throughput imaging of protein crystallization plates. It allows for efficient monitoring of crystal growth by incubating and capturing high-quality images of up to 1,000 microplates based on a user-defined schedule.
This system reduces the need for manual inspection under a microscope, ensuring consistent and precise imaging over extended periods. With its temperature-controlled environment and user-friendly interface, ROCK IMAGER 1000 facilitates the identification of crystal formation and optimization of crystallization conditions.

Formulator, Formulatrix
The FORMULATOR robot is designed to optimize protein crystallization conditions based on initial screening results.
This advanced system refines successful crystallization parameters to enhance crystal quality and growth. The FORMULATOR excels in its ability to mix up to 16 different solutions with precise volume control, creating a diverse array of customized formulations. It then dispenses these optimized solutions onto appropriate plate formats, streamlining the crystallization process. This capability allows researchers to efficiently explore a wide range of conditions around promising initial hits, significantly increasing the likelihood of obtaining high-quality crystals suitable for structural studies.
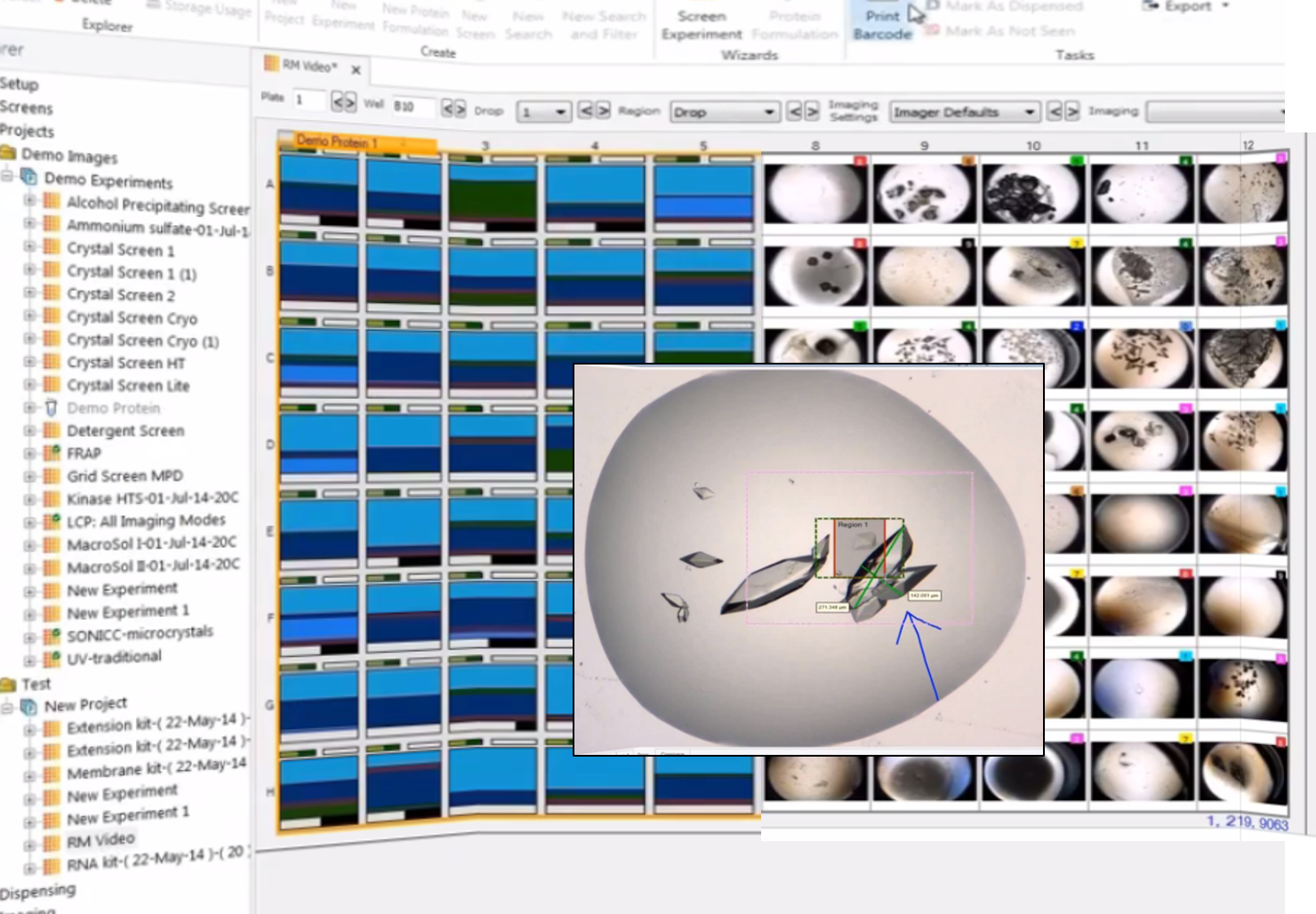
Rock Maker Software
Rock Maker Software is a comprehensive solution designed to manage and streamline the entire protein crystallization process. This powerful yet user-friendly software serves as a central hub for crystallization experiments.
Rock Maker offers a range of advantages, including the ability to control experiment design, automate image scheduling for the Rock Imager system, and provide a straightforward interface for examining and scoring crystallization drops. Additionally, it seamlessly integrates with other crystallization tools, allowing users to design optimized screens for the Formulator robot. This integration and versatility make Rock Maker an essential tool for researchers seeking to efficiently conduct and analyze protein crystallization experiments.
X-ray Data Collection
Protein crystal handling and X-ray data collection are critical steps in the process of determining the three-dimensional structures of proteins.
Once high-quality protein crystals are obtained, they must be carefully mounted and freezed before X-ray diffraction experiments. This process involves precise manipulation of the crystals, followed by their rapid freezing to protect them from radiation damage during data collection.

Discovery V20 stereo microscope, Zeiss
The Zeiss Discovery V20 stereo microscope simplifies the delicate task of crystal mounting with its advanced features.
Its motorized 20x zoom capability allows researchers to precisely inspect and manipulate protein crystals.

X-ray data collection
Once the protein crystals are frozen in liquid nitrogen, they are shipped to synchrotron facilities like the European Synchrotron Radiation Facility (ESRF) for high-resolution X-ray diffraction analysis.
The ESRF's intense synchrotron radiation, coupled with streamlined access through the Israeli Block Allocation Group (BAG), provides researchers with efficient, priority access to state-of-the-art technology. This arrangement significantly accelerates data collection and enhances the quality of structural information obtained, ultimately speeding up scientific discoveries in the field of structural biology.
Data Analysis and Computation
Data analysis and computation in protein X-ray crystallography involve converting X-ray reflection data measured on the detector into usable intensity values.
This process requires advanced software for processing and phase determination, essential for solving the protein structure. Tools like HKL3000 are commonly used to streamline these tasks, automating data reduction, scaling, and phasing. In our Graphics room, iMac workstations with high-performance graphics capabilities enable efficient computation and molecular visualization, making them ideal for modeling protein structures.